The MassXpert software (currently MassXpert3) is a linear polymer chemistry modelling program that is aimed at simulating mass spectrometric data for (bio) polymeric sequences: once a polymer chemistry definition has been modelled, a polymer sequence may be entered in the editor and a large number of polymer chemistry reactions might be simulated along with theoretical mass spectrometric data.
MassXpert comprises the following four modules
XpertDef
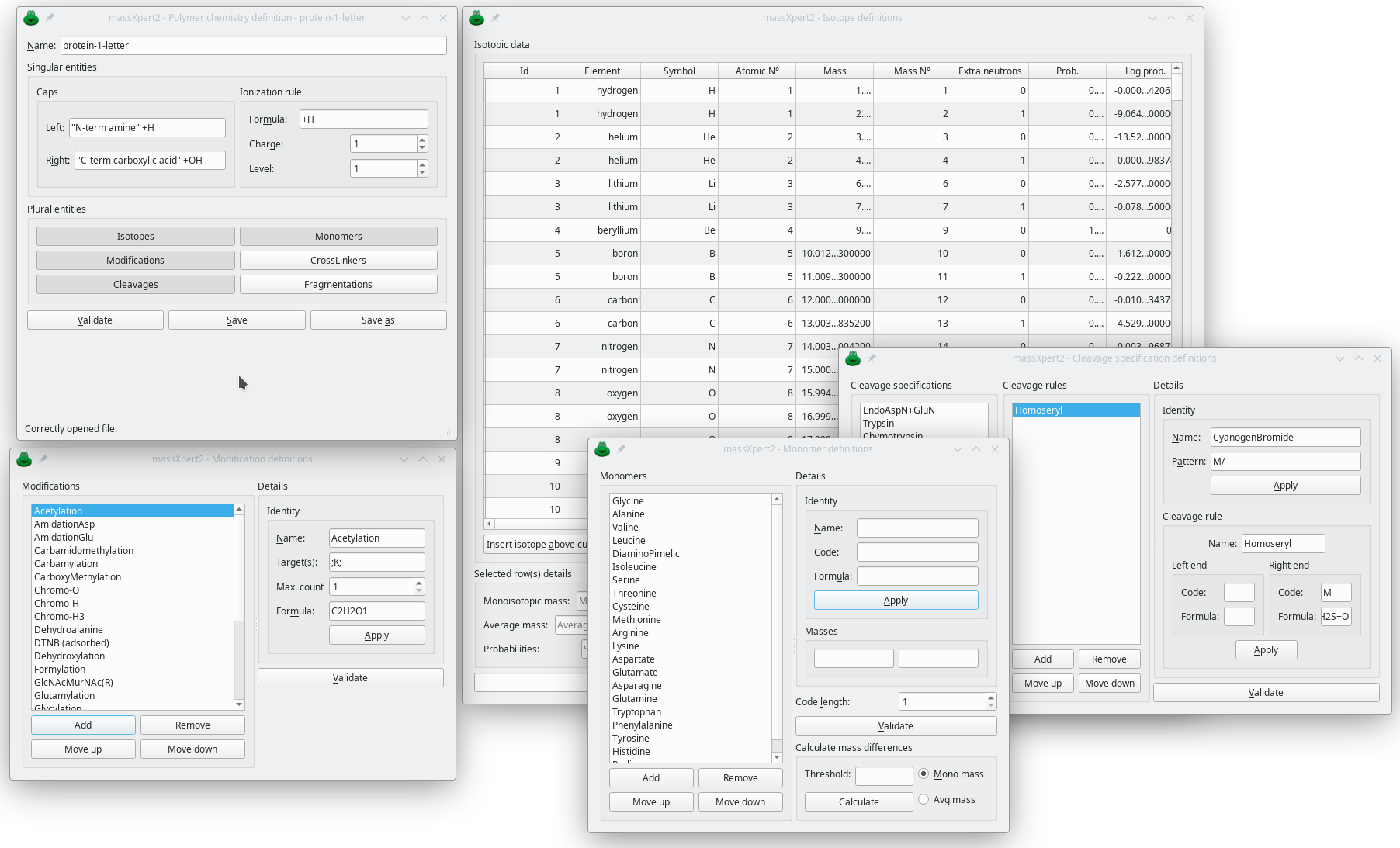
With this module the user defines brand new polymer chemistries. At the very least, a polymer chemistry comprises the following chemical informations:
-
A table of isotopic data (isotopes needed to describe chemical elements)
-
A list of monomers defined as a code (any number of letters), a name and a formula. The formula defines the residue, not the monomer.
-
A list of (bio) chemical modifications defined as a name, a net chemical formula (the net atomic differential between the unmodified and modified states), a list of monomer targets that might be modified with this modification, and finally the maximum number of such modifications that can occur on a single monomer target (for methylation, that would be 3, for example, in histones; or for phosphorylation, that would be 1, for the threonine, serine, tyrosine residues).
-
A list of cleaving agents (either chemical or biochemical), like proteolytic enzymes or endonucleolytic enzymes, but also like cyanogen bromide. The flexibility of the grammar used to define the modifications allows for the definition of monomer modifications occurring when using chemical reagents like cyanogen bromide, for example, where the methionyl residue is converted to a homoseryl residue.
-
A list of gas phase fragmentation patterns for the detailed simulation of precursor ion fragmentation of the matching polymer definition. The grammar used for the definition of gas phase fragmentation patterns is that flexible that it allows the description of the most complex fragmentation mechanisms in the saccharidic world.
-
The polymer sequence caps. In the peptide/protein world, the polymer is a chain of monomer residues that, in order to correspond to a finished polymer sequence, needs to be capped with both a proton at the N-terminal end and a hydroxyl group at the C-terminal end.
-
The default ionization rule to be used when computing the elemental composition and the masses of the polymer or of the oligomer (protein or peptide, for example). An ionization rule is defined by specifying the chemical modification that the analyte is subjected to during the ionization, the charge that is brought by that ionization reaction and finally the number of times the ionization reaction is to be performed onto the analyte. For MALDI mass spectrometry of proteins, typically, that would be [+H, 1, 1] which reads like “Protonation brings one proton to the analyte, thus bringing a charge of one a single time”.
XpertCalc
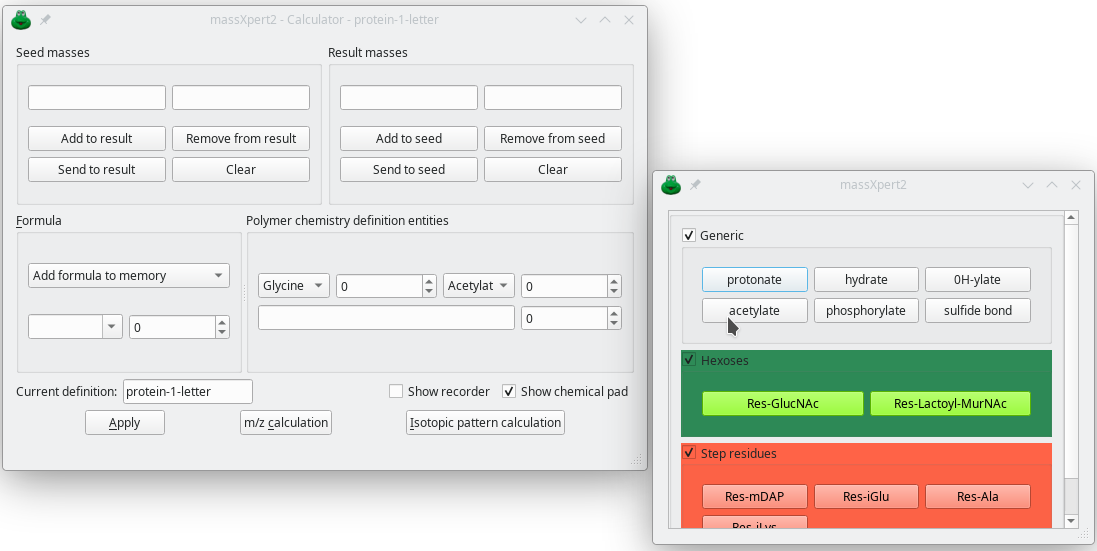
With this module, you get a desktop calculator that understands your polymer chemistry definitions as defined in XpertDef. The calculator allows any kind of chemical reaction and is infinitely programmable. A chemical pad is available in which chemical reactions previously defined become available in the form of buttons, exactly like with a desktop calculator. Any calculation is recorded in a logbook that is exportable to put in the lab-book.
XpertEdit
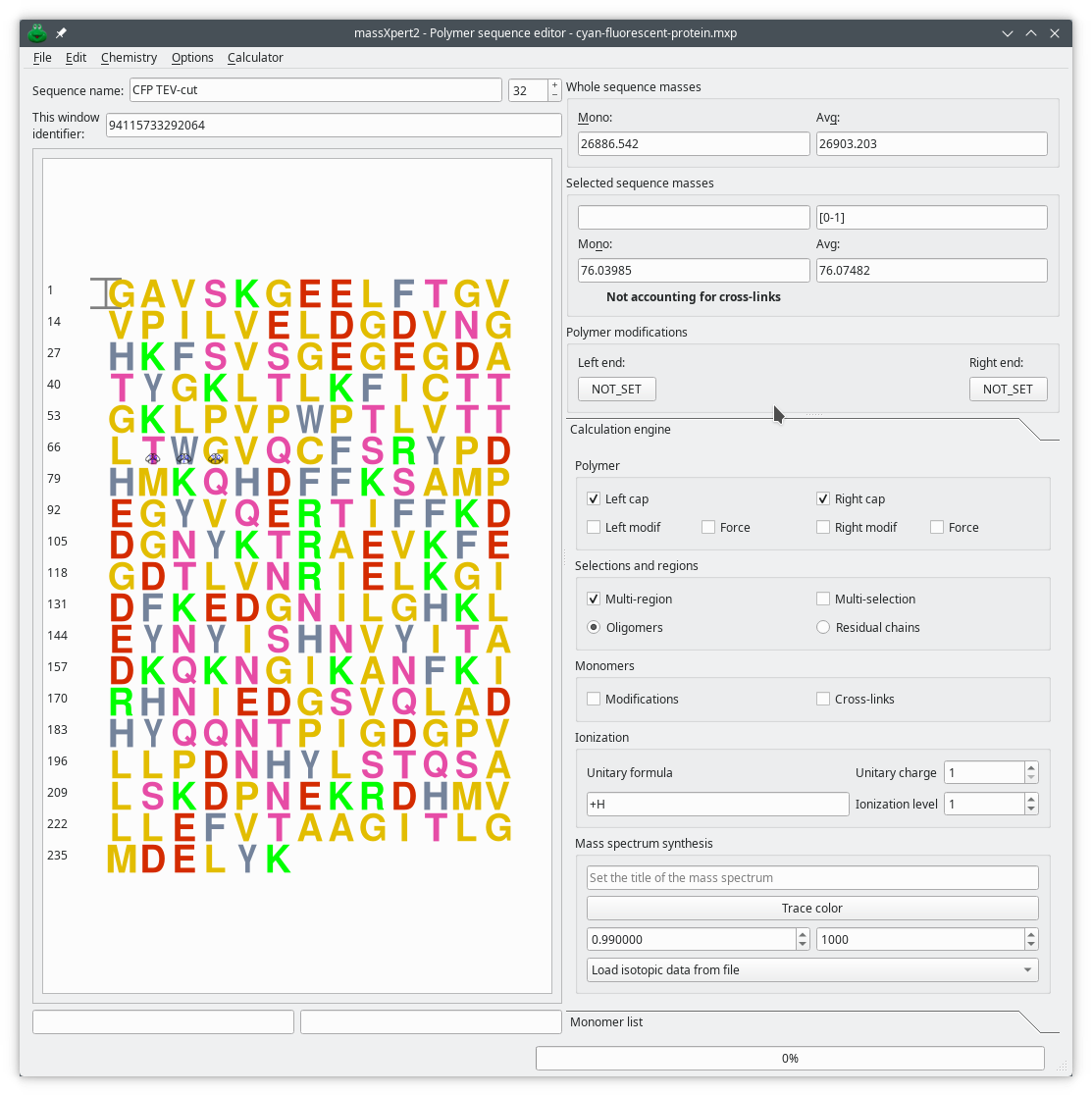
With this module, you get a sophisticated polymer sequence editor and a chemical center where a huge amount of simulations might be performed. Anything mass-related is virtually feasible in XpertEdit. The polymer sequence editor is configurable to the extent that the graphical representation of the monomer residues in the sequence is definable using a drawing program that outputs SVG files (like Inkscape, for example).
Isotopic cluster simulation
Simulating isotopic clusters from a formula — traditionally a fairly complex task — is made easy thanks to the interface available in MassXpert. You simply need to enter the formula of the analyzed molecule (adding the number of ionization agents — typically protons when analyzing proteins):
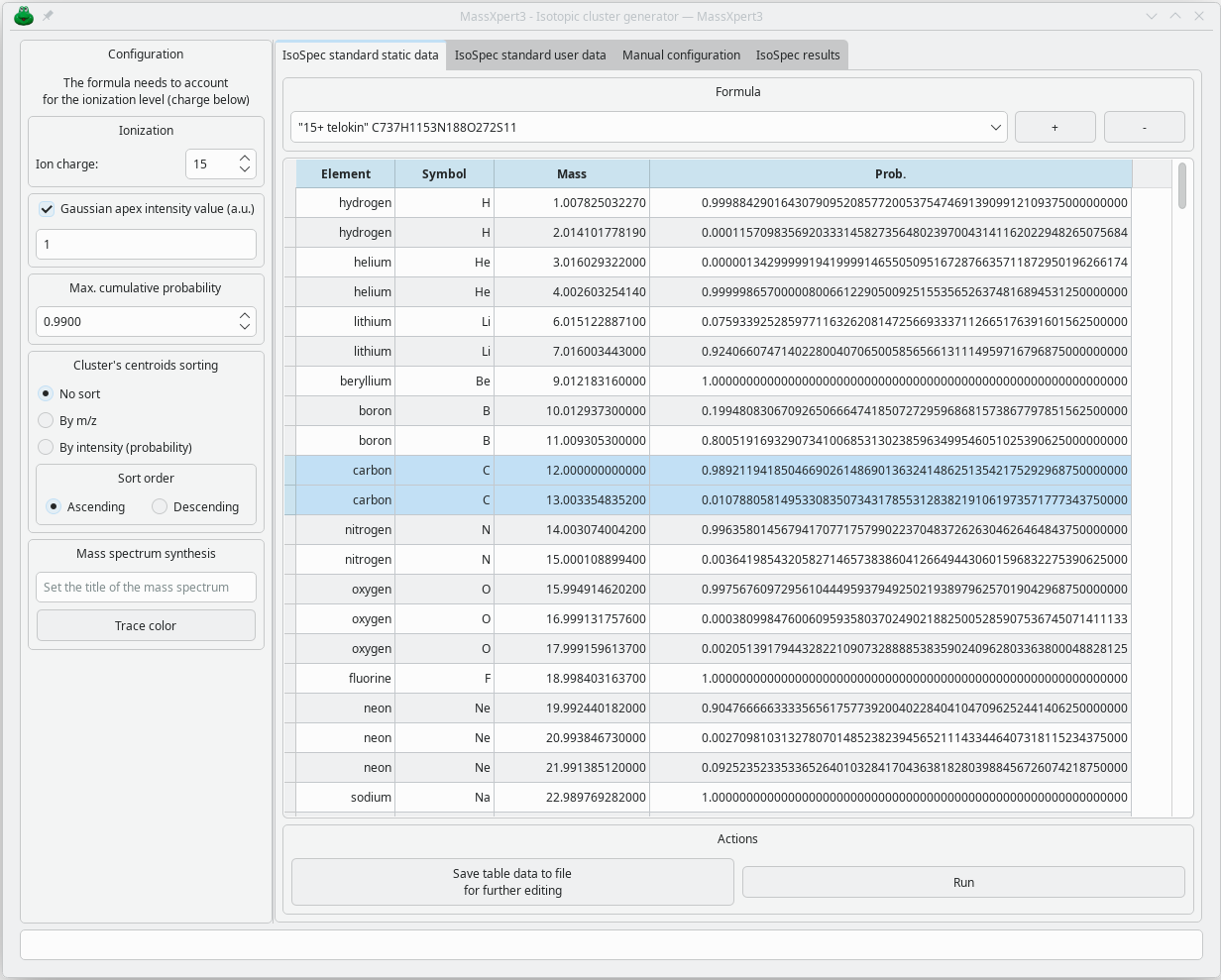
Once this configuration is done, the isotopic cluster is presented as a histogram whose x-values are the m/z values and whose y-values are the probabilities. However, these values are only centroids and do not provide an idea of the appearance of the isotopic cluster, whose profile depends on the resolving power of the instrument. To obtain the correct profile, you must use the functionality illustrated below:
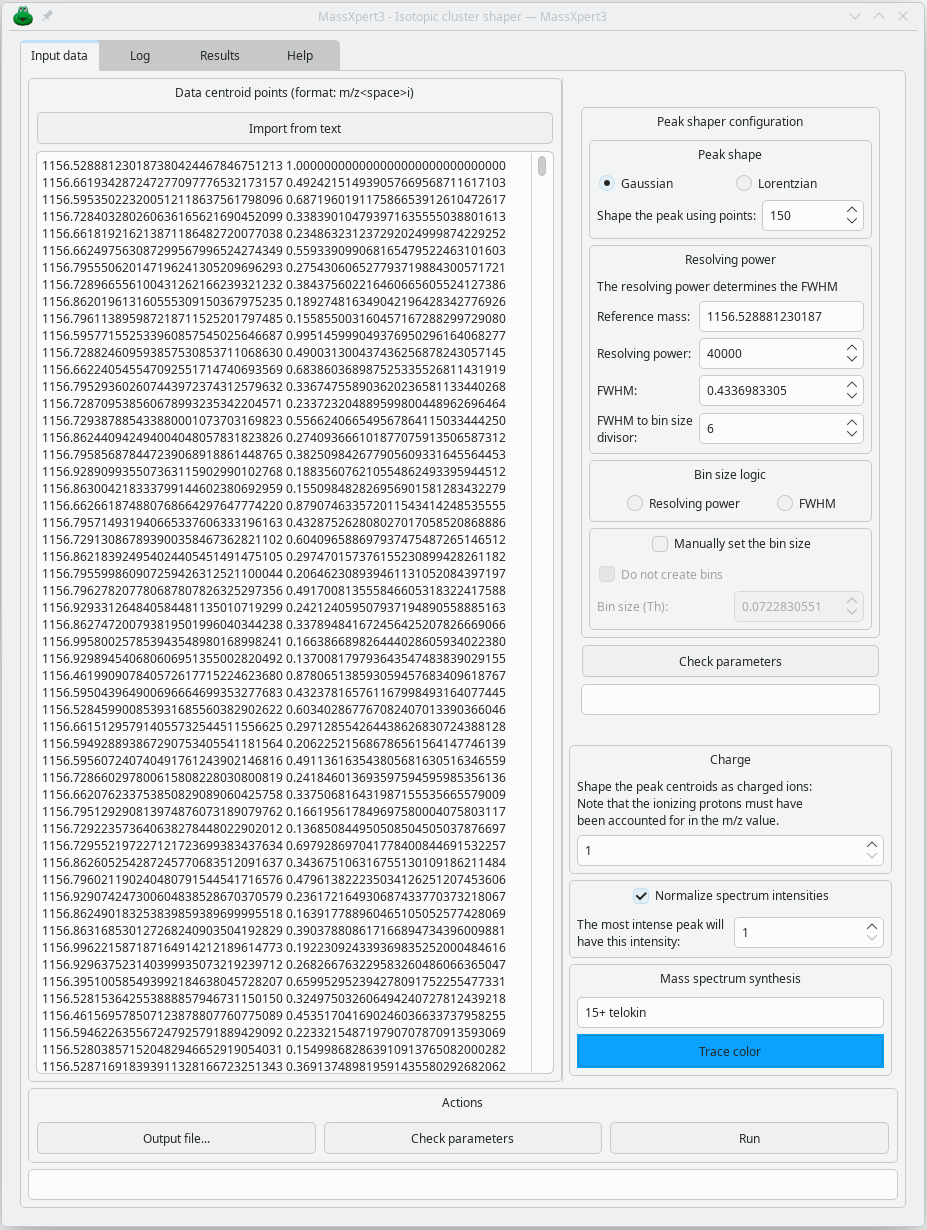
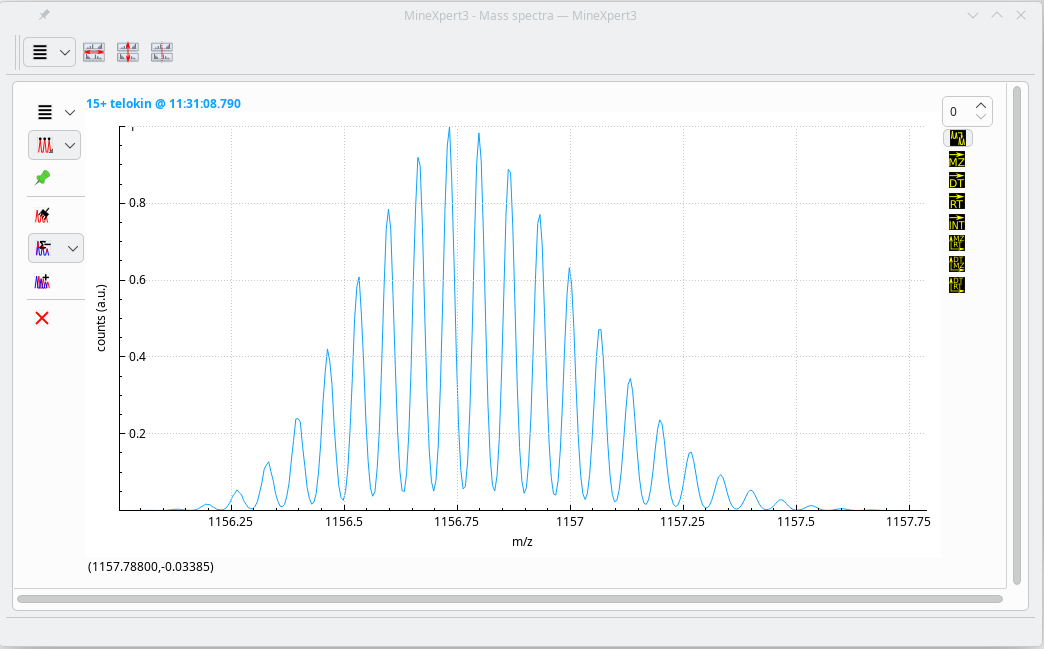
Once the spectrum has been generated as it would be by a mass spectrometer with a given resolving power, it is displayed in the MineXpert3 program:
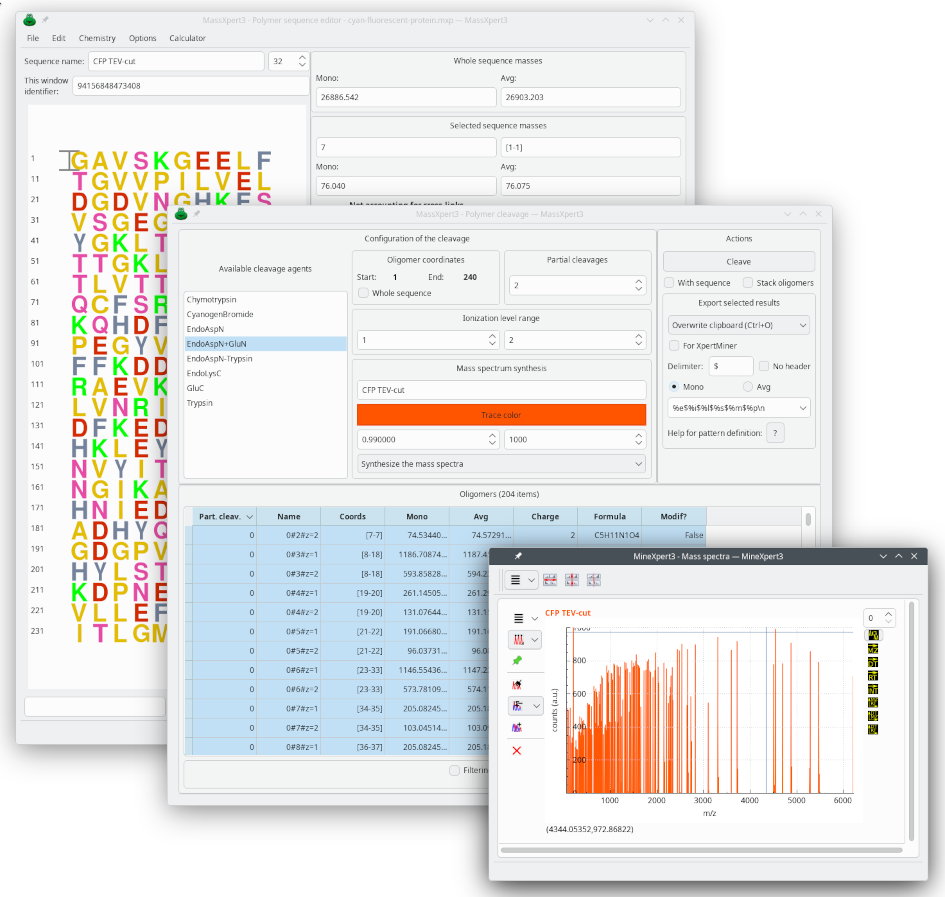
Cleavage and mass spectrum simulation
When the polymer sequence is ready in the sequence editor, it is possible, among many other options, to hydrolyze it with the enzyme (or enzyme cocktail) of your choice. The oligomers can then be used to simulate a mass spectrum of the hydrolysate, based on the instrument’s resolving power. This is illustrated in the figure below, where the synthetic mass spectrum has automatically been displayed in MineXpert:
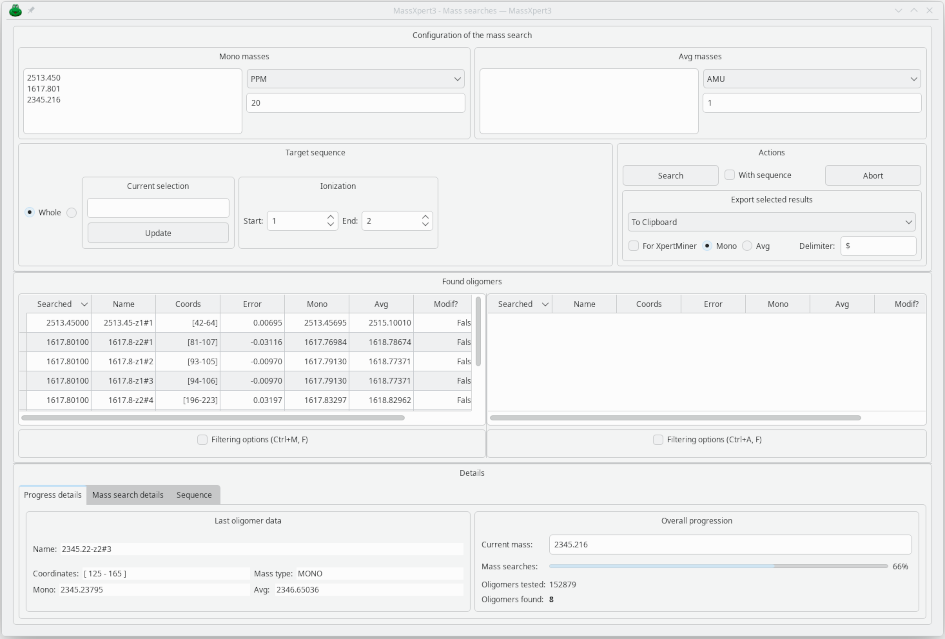
Arbitrary Mass Searches
It is possible to perform mass (or m/z) searches on a given sequence. Typically, this is useful when certain mass peaks cannot be explained by a specific enzymatic digestion.
JavaScript
With the JavaScript module, you have a data-scripting environment in which most of the features of MassXpert are accessible from the ECMAScript environment.
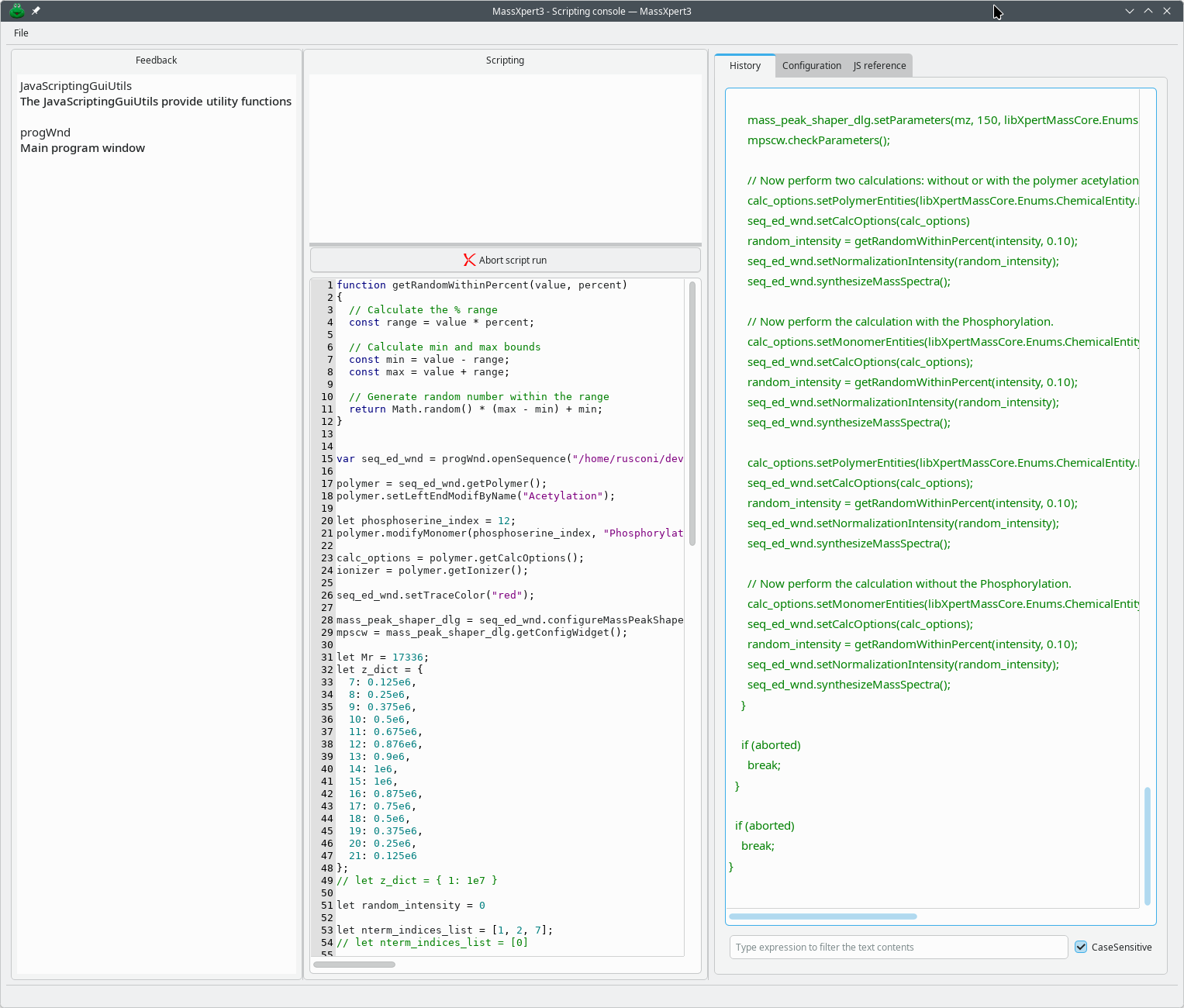
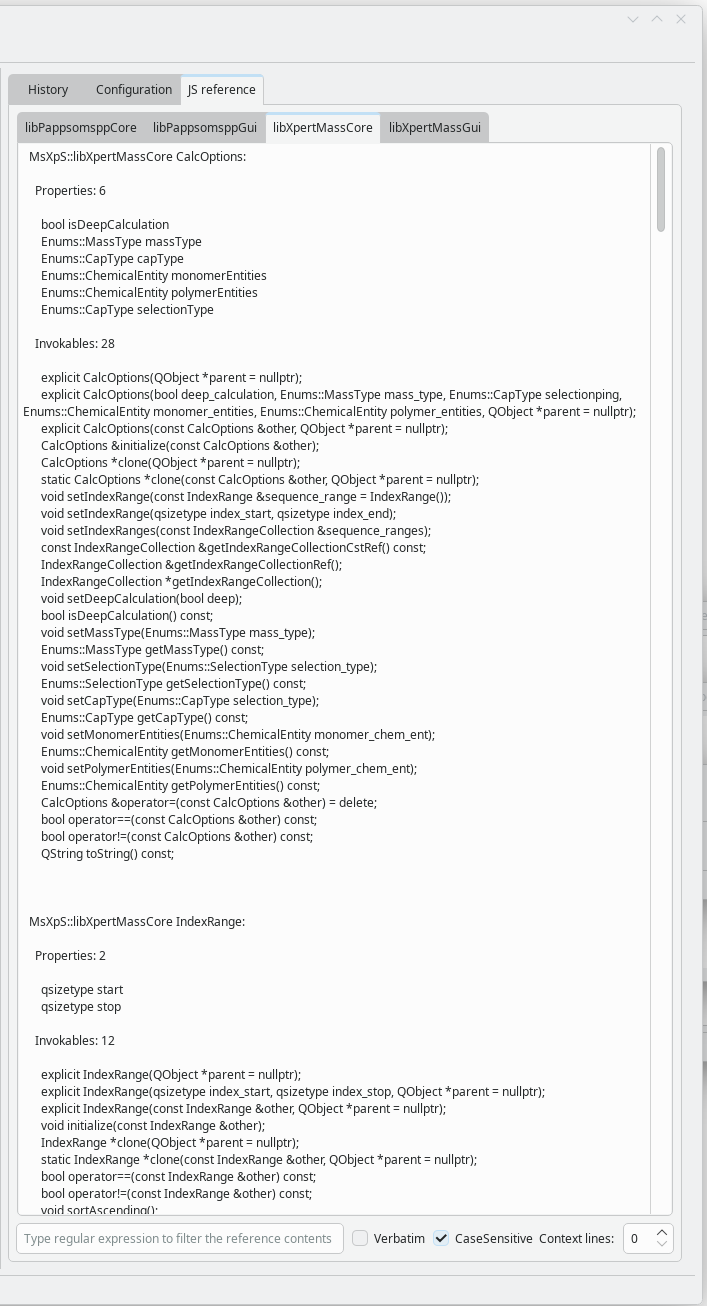
The JavaScript documentation is extracted from dedicated tags in the source code and is therefore reliable. It is displayed for each component (particularly the libraries) in the JS Reference tab, as shown below: